Markus Strasser
Powered by 🌱Roam GardenNatural Language Understanding of Scientific Research: Ecosystem, Datasets, Tools, Problems
Grave of the fireflies Recommended Movies
TODO: group, clean it up and put most things in table format
This is a private list and accordingly chaotic
natural language parsing and information extraction
Stanford CoreNLP and their [information extraction]. Available in python through spacy + stanza
BioStanza paper: Stanza added NER support for biomedical, beating
[[interval:175.0]] [[factor:2.60]] March 1st, 2021
Stanza: Multilingual, fully neural NLP processing
spaCy code snippets and examples including information extraction
AllenAI Science Parse specifically for scholarly work. Status: not actively developed anymore
Grobid for PDF parsing
NLP Platforms, Libraries and algorithm
[[interval:1.4]] [[factor:2.15]] October 12th, 2020
Textattack - util for adversarial attacks on NLP models
SyntaxGym Evaluating Language Models as psycholinguistic subjects - MIT Computational Psycholinguistics Laboratory
Med7 Model. Amazing NER for clinical data. colab
[[interval:107.2]] [[factor:2.65]] March 13th, 2021
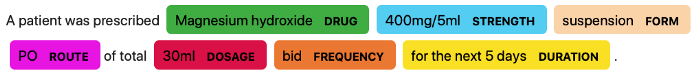
medaCy is a text processing and learning framework built over spaCy to support the lightning fast prototyping, training, and application of highly predictive medical NLP models.
from medacy.model.model import Model
model = Model.load_external('medacy_model_clinical_notes')
annotation = model.predict("The patient was prescribed 1 capsule of Advil for 5 days.")
print(annotation)
When an area is exploding in options, like NLP, not expertise, but faster feedback cycles and creativity win the game
Prodigy from the makers of Spacy
Prodigy is a fully scriptable annotation tool so efficient that data scientists can do the annotation themselves
Thinc: functional and typed deep learning
Spacy Course
Frontends with Python
FastAPI is a modern, fast (high-performance), web framework for building APIs
Typer: for CLI API development
Neural Constituency Parsing. More advanced implementation in AllenNLP
whatlies embedding data visualization interactive. Also PCA, umap. Integrates nice with spacy

Sentence-Transformers, also through spaCy
This repository fine-tunes BERT / RoBERTa / DistilBERT / ALBERT / XLNet with a siamese or triplet network structure to produce semantically meaningful sentence embeddings that can be used in unsupervised scenarios: Semantic textual similarity via cosine-similarity, clustering, semantic search.
NegBio uncertainty, negation and hedge cue detection in clinical reports
Epitator spacy pipeline component: adds epidemic annotations to text
dependency forest: differentiable parser
Dependency Forest
excerpt::
A dependency forest encodes all valid dependency trees of a sentence into a 3D space that syntax parsing is differentiable
We proposed an efficient and effective relation extraction model that leverage full dependency forests, each of which encodes all valid dependency trees into a dense and continuous 3D space.
we define a full forest as a 3-dimensional tensor, with each point representing the conditional probability p(wj , l|wi) of one word wi modifying another word wj with a relation l.
Compared with a 1-best tree, a full dependency forest efficiently represents all possible dependency trees within a compact and dense structure, containing all possible trees (including the gold tree).
This method allows us to merge a parser into a relation extraction model so that the parser can be jointly updated based on end-task loss.
Extensive experiments show the superiority of forests for RE, which significantly outperform all carefully designed baselines based on 1-best trees or surface strings.
HuggingFace: Democratize NLP
spaCy and Cython for 100x faster processing. Note: most spacy classes already use Cython
Spacy integrated co-reference resolution: NeuralCoref
SciSpacy Demo shows how Linking and NER works on Streamlit
SciSpacy has different NER recognition models depending on biomedical subdomain (drug/disease, cancer, genomics)
Longformer for Bert long documents with gradient checkpointing ++
SciRex: Dataset for Document Level text extraction
OpenRE: relation extraction with confidence score
excerpt::
TL;DR: More efficient text encoders emerge from an attempt (failed?) to do "GAN's for Text".
The authors “train a model on one GPU for 4 days that outperforms GPT (trained using 30x more compute) on the GLUE natural language understanding benchmark.” (Clark et al., 2020)
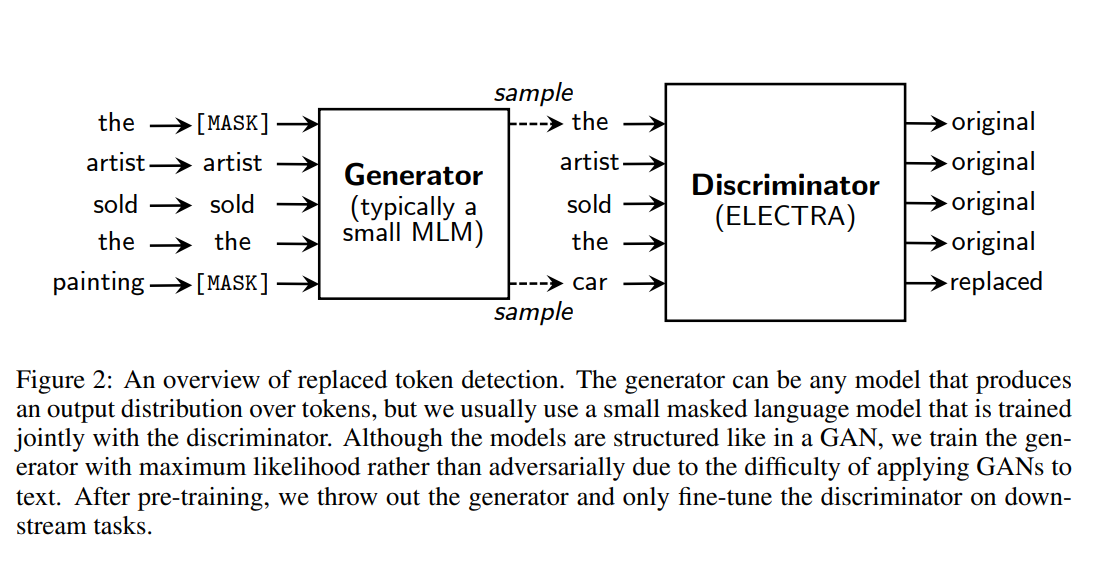

Going Binary improves sample efficiency: Instead of [MASKED] token, the Discriminator only has to predict if a viable swap was sampled from a generator (painting -> mask -> car). Later the generator is thrown away
__more sample-efficient pre-training task called replaced token detection. Instead of masking the input, our approach corrupts it by replacing some tokens with plausible alternatives sampled from a small generator network.
Then, instead of training a model that predicts the original identities of the corrupted tokens, we train a discriminative model that predicts whether each token in the corrupted input was replaced by a generator sample or not.
more efficient than MLM because the task is defined over all input tokens rather than just the small subset that was masked out. __
30x training efficiency over BERT
BioMedical Claim Extraction: With Code and AWS and PubMed setup. Precision: 0.88. Paper comes with dataset with 1500 annotated abstracts
Jeff Hawkin's Numena uses a very different approach for cognition
SciFact: claim verification; evaluate veracity of scientific claims. Double digit improvements
[[interval:2.4]] [[factor:2.60]] January 5th, 2021
In Scifact a claim is an atomic verifiable statement
Interesting errors and work to be done:

Science background includes knowledge of domain-specific lexical relationships – e.g. Drosophila is a synonym for fruit fly.
Directionality requires understanding increases or decreases in scientific quantities – e.g. decreased (but not increased) lipogenesis implies impairment of the lipogenesis process.
Numerical reasoning involves interpreting numerical or statistical statements, often reporting confidence intervals or p-values.
causality and effect requires reasoning about counterfactuals – e.g. if knocking out MVP inhibits tumor progression, then the presence of MVP enables tumor growth and aggression
Trained Models
dataset/bio/description::
We release the dataset of annotated 1,500 abstracts containing 11,702 sentences (2,276 annotated as claim sentences) sampled from 110 biomedical journals.
dataset/bio/comment::
interoperability Efforts
HierPlane Visualize Tree Structures Linguistic Visualization
[[interval:274.6]] [[factor:2.95]] September 22nd, 2021
UMLS
The UMLS integrates and distributes key terminology, classification and coding standards, and associated resources to promote creation of more effective and interoperable biomedical information systems and services, including electronic health record
Challenges, Methods, Tasks
Hedge cue detection: detecting uncertain or hypothetical statements vs definite statements
Claim detection, argument mining and finding supporting or contradicting statements
dataset, corpi and benchmark; most annoted
Initially I thought the problem is great datasets, but now I think it's finding out which of the 100s of corpi are easiest to parse and build on
dataset/bio/description::
Dependencies, genetic, cancer etc.
dataset/bio/comment::
INDRA uses some of it for DepMap Explainer
dataset/bio/description::
dataset/bio/comment::
annotated. 1500~ sentences
dataset/bio/description::
Our model-based candidates are iteratively updated to contain more difficult negative samples as our model evolves. In this way, we avoid the explicit pre-selection of negative samples from more than 400K candidates. On four biomedical entity normalization datasets having three different entity types (disease, chemical, adverse reaction), our model BIOSYN consistently outperforms previous state-of-the-art models almost reaching the upper bound on each dataset.
dataset/bio/comment::
Seems popular for very short lifespan
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
Grounding Protein Relations (isa, part-of)
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
50GB FTP
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
dataset and pipeline for entailment recognition
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
MIMIC-III (Medical Information Mart for Intensive Care III) is a large, freely-available database comprising deidentified health-related data associated with over forty thousand patients who stayed in critical care units of the Beth Israel Deaconess Medical Center between 2001 and 2012.
The database includes information such as demographics, vital sign measurements made at the bedside (~1 data point per hour), laboratory test results, procedures, medications, caregiver notes, imaging reports, and mortality (both in and out of hospital).
dataset/bio/comment::
Causal BioNet CBN
dataset/bio/description::
dataset/bio/comment::
Private BEL databases can be licensed from Qiagen Ingenuity, Clarivate etc.
The Causal Biological Networks (CBN) database is composed of multiple versions of over 120 modular, manually curated, BEL-scripted biological network models supported by over 80,000 unique pieces of evidence from the scientific literature.
They represent causal signaling pathways across a wide range of biological processes including cell fate, cell stress, cell proliferation, inflammation, tissue repair and angiogenesis in the pulmonary and vascular systems.
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
CancerMine, a text-mined and routinely updated database of drivers, oncogenes and tumor suppressors in different types of cancer. All data are available online
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
Entity Normalization and recognition works well. Downloadable in full via FTP! ...30GB yumyum!
dataset/bio/description::
S2ORC is a large contextual citation graph of English-language academic papers from multiple scientific domains; the corpus consists of 81.1M papers, 380.5M citation edges, and associated paper metadata. We provide structured full text for 8.1M open access papers.
All inline citation mentions in the full text are detected and linked to their corresponding bibliography entries, which are linked to their referenced papers, forming contextual citation edges. To our knowledge, this is the largest publicly-available contextual citation graph. The full text alone is the largest structured academic text corpus to date.
dataset/bio/comment::
Used
License tricky for commercial products
dataset/bio/description::
Never understood what this was supposed to be
And as always when the EU tries to coordinate: total failure
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
dataset/bio/description::
Biomedical Language Understanding Evaluation benchmark
BLUE benchmark consists of five different biomedicine text-mining tasks with ten corpora.
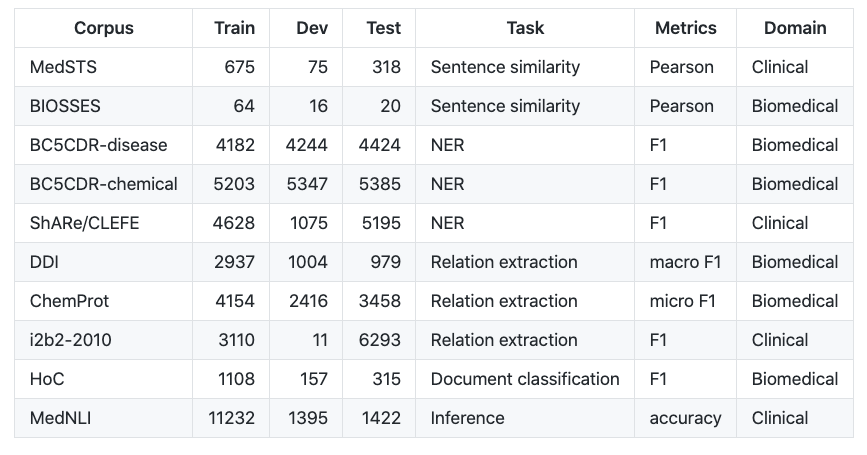
dataset/bio/comment::
Amazing collection
DDI (Drug-Drug Interaction) corpus
NacTem corpi 💫
dataset/bio/description::
links to corpora of various sizes, with different levels of annotation, and belonging to different domains.
dataset/bio/comment::
SNOMED CT ontology
BioNLP data NCBI
dataset/bio/description::
collection resources
dataset/bio/comment::
🌲 🏦 Treebanks, Treebanks, Treebanks!
Pubmed 200k RCT: a Dataset for Sequential Sentence Classification in Medical Abstracts](https://github.com/Franck-Dernoncourt/pubmed-rct)
dataset/bio/description::
The dataset consists of approximately 200,000 abstracts of randomized controlled trials, totaling 2.3 million sentences. Each sentence of each abstract is labeled with their role in the abstract using one of the following classes: background, objective, method, result, or conclusion.
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::
unsuspected
dataset/bio/description::
lots of good training data and compound library
dataset/bio/comment::
BioRel Corpus
dataset/bio/description::
We present BioRel, a large-scale dataset constructed by using Unified Medical Language System (UMLS) as knowledge base and Medline as corpus. Entities in sentences of Medline are identified and linked to UMLS by Metamap. Relation label for each sentence is recognized using distant supervision.
dataset/bio/comment::
dataset/bio/description::
The AImed corpus consists of 225 Medline abstracts. 200 abstracts describe interactions between human proteins, 25 do not refer to any interaction. There are 4084 protein references and around 1000 tagged interactions in this data set. In this data set there is no distinction between genes and proteins and the relations are symmetric.
dataset/bio/comment::
dataset/bio/description::
dataset/bio/comment::

dataset/bio/description::
Another Dataset for causal tagging
dataset/bio/comment::
🎉 List of biomedical datasets and tools
dataset/bio/description::
Great Collection
dataset/bio/comment::
DDI, PGR, and BC5CDR corpus
dataset/bio/description::
well known corpus
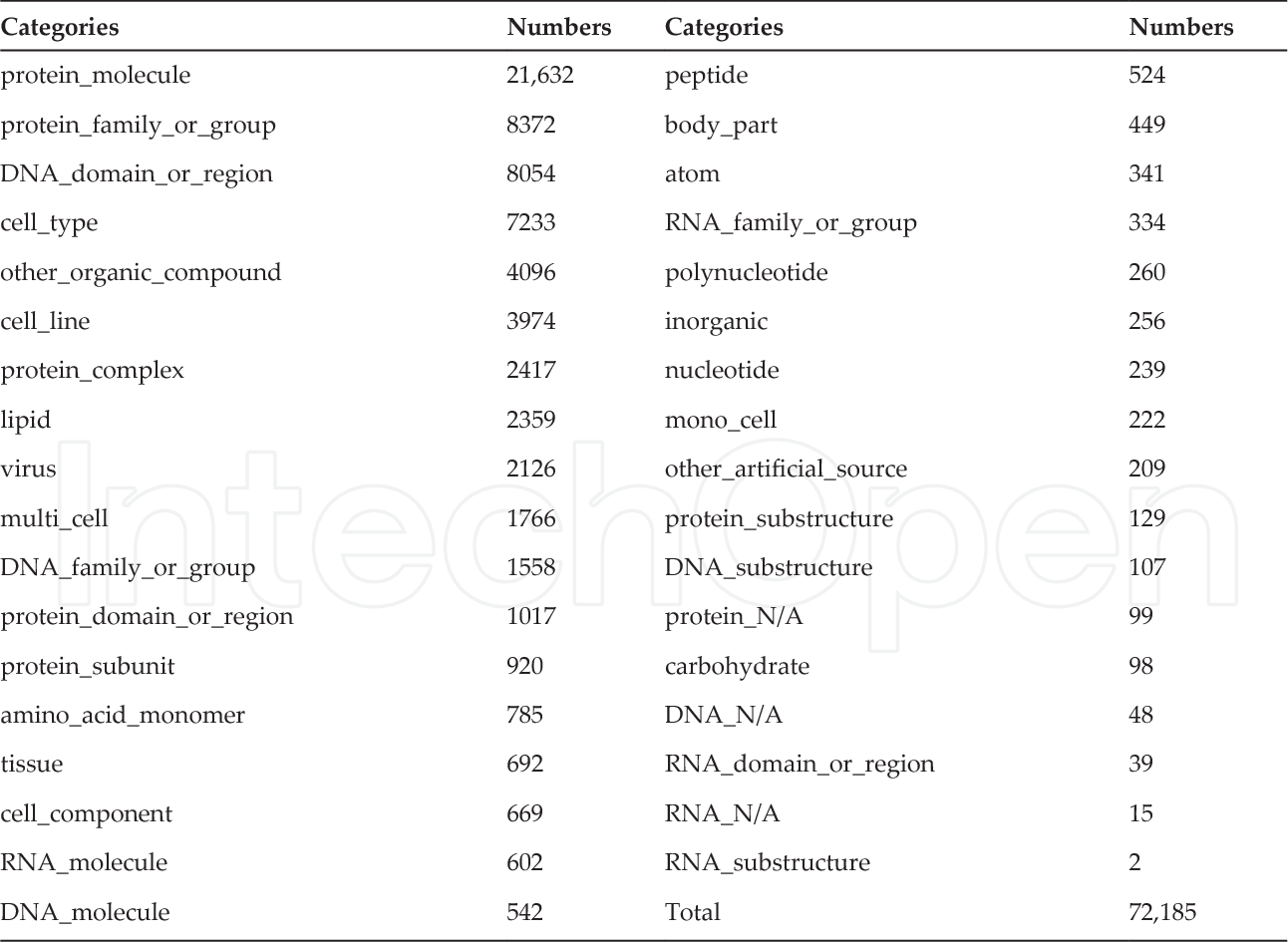
dataset/bio/comment::
"We evaluate our tLSTM model on five publicly available PPI corpora: AIMed 37(https://scite.ai/reports/10.1016/j.artmed.2004.07.016), BioInfer 38(), IEPA 39(https://scite.ai/reports/10.1142/9789812799623_0031), HPRD50 40(https://scite.ai/reports/10.1093/bioinformatics/btl616) and LLL [41]"
dataset/bio/description::
dataset/bio/comment::
Very structured but unfortunately no set for biomedicine
excerpt::
Biomedical Relations: THEMES (possible relation types)
chemical-gene
(A+) agonism, activation
(A-) antagonism, blocking
(B) binding, ligand (esp. receptors)
(E+) increases expression/production
(E-) decreases expression/production
(E) affects expression/production (neutral)
(N) inhibits
gene-chemical
(O) transport, channels
(K) metabolism, pharmacokinetics
(Z) enzyme activity
chemical-disease
(T) treatment/therapy (including investigatory)
(C) inhibits cell growth (esp. cancers)
(Sa) side effect/adverse event
(Pr) prevents, suppresses
(Pa) alleviates, reduces
(J) role in disease pathogenesis
disease-chemical
(Mp) biomarkers (of disease progression)
gene-disease
(U) causal mutations
(Ud) mutations affecting disease course
(D) drug targets
(J) role in pathogenesis
(Te) possible therapeutic effect
(Y) polymorphisms alter risk
(G) promotes progression
disease-gene
(Md) biomarkers (diagnostic)
(X) overexpression in disease
(L) improper regulation linked to disease
gene-gene
(B) binding, ligand (esp. receptors)
(W) enhances response
(V+) activates, stimulates
(E+) increases expression/production
(E) affects expression/production (neutral)
(I) signaling pathway
(H) same protein or complex
(Rg) regulation
(Q) production by cell population
dataset/bio/description::
manually annotated.
Relationship Types
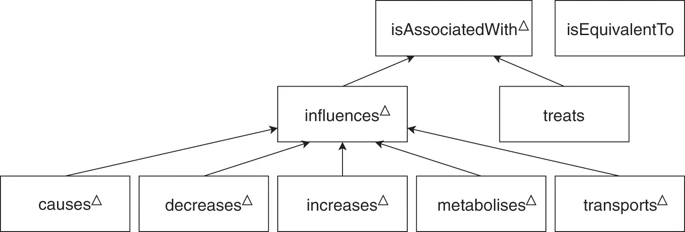
dataset/bio/comment::
dataset/bio/description::
“What distinguishes MedMentions from other annotated biomedical corpora is its size (over 4,000 abstracts and over 350,000 linked mentions), as well as the size of the concept ontology (over 3 million concepts from UMLS 2017) and its broad coverage of biomedical disciplines”
dataset/bio/comment::
ezTag
To support annotating a wide variety of biological concepts with or without pre-existing training data, we developed ezTag, a web-based annotation tool that allows curators to perform annotation and provide training data with humans in the loop. ezTag supports both abstracts in PubMed and full-text articles in PubMed Central. It also provides lexicon-based concept tagging as well as the state-of-the-art pre-trained taggers such as TaggerOne, GNormPlus and tmVar. ezTag is freely available at http://eztag.bioqrator.org
dataset/bio/description::
A collections of public and free annotated datasets of relationships between entities/nominals
dataset/bio/comment::
Data programming is a paradigm that circumvents this arduous manual process by combining databases with simple rules and heuristics written as label functions, which are programs designed to automatically annotate textual data.
Unfortunately, writing a useful label function requires substantial error analysis and is a nontrivial task that takes multiple days per function. This makes populating a knowledge graph with multiple nodes and edge types practically infeasible.
Data Programming for fast creation annotated training sets
We therefore propose a paradigm for the programmatic creation of training sets called data programming in which users express weak supervision strategies or domain heuristics as labeling functions, which are programs that label subsets of the data, but that are noisy and may conflict.
We show that by explicitly representing this training set labeling process as a generative model, we can "denoise" the generated training set, and establish theoretically that we can recover the parameters of these generative models in a handful of settings
WebAnno annotation tool
Iterative Annotation
by switching to an approach where the human can continuously improve the model by annotation while using the model to extract information, with the especially good news that the largest model improvements are achieved already very early in the process, as long as the domain is confined.

CiteSpace Scientometric visualization
CiteSpace has three core concepts: burst detection, betweenness centrality, and heterogeneous networks. These concepts can solve three practical problems: identifying the nature of research frontiers, marking keywords, and identifying emerging trends and sudden changes in time.
The main procedural steps of CiteSpace software are time slicing, thresholding, modeling, pruning, merging, and mapping and the main source of input data for CiteSpace is the Web of Science database. CiteSpace can identify frontier areas of current research by extracting burst terms from identifiers of titles, abstracts, descriptors, and bibliographic records. CiteSpace also makes it easier for users to recognize key points by identifying nodes with high betweenness centrality
benchmark and metric and evalution
r/Doc
SemEval 2020 Tasks, like counterfactual identification
BioRelEx 2000 annotated sentences
NELeval (named entity linking)
Cohen's Kappa: measures annotator agreement
Formats, Utils and DSLs: Write less code
brat standoff format - .ann comes with utils like merging .ann with .txt and parsing OBO ontology data. Of course written in horribly-imperative-hardly-readable-3forloopsdeep python code. Ever heard of composition?
Custom spaCy pipeline components can save much of machine learning work eg.
Example Code and Projects
Check every month or so: https://huggingface.co/models
[[interval:321.3]] [[factor:3.25]] November 8th, 2021
Literature Based Discovery: Lion Cambridge Cancer Effort 22GB dataset
dataset/bio/description::
List of resources and datasets!
dataset/bio/comment::
[[interval:114.5]] [[factor:2.80]] March 19th, 2021
EvidenceMiner includes also a web search interface that's quite OK for an academic project
PubRunner: run tools against PubMed releases
An AllenAI researcher made a colab with sciSpacy
that explores
Rethinking Relationship Types Semantic Role Labeling | SRL
producer - consumer, cause - effect, container - content, inhibitor - inhibited
People, Groups, Conferences
SIGIR information extraction conference
Jake Lever: did Kindred for biomedical text mining in spacy. His Papers
Mark Neumann (@AllenAI)
BioASQ organizes challenges. Plus has wordvecs trained on 10M Pubmed articles.
Vatlab SoS data analysis environment
Observations
There should be a quality control and independent ranking scheme and website for biomedical datasets akin to what labdoor is for supplements
The UID or DOI of each dataset should let you search for corresponding data crunching algorithms or notebooks with an EDA
1% of people that use python have heard about composition
Upcoming Tasks
No matter how fast ML advances, people stay unsatisfied. That's good because datasets, benchmarks and algorithms have to incorporate higher and higher level features of discourse
TODOs:
Extract Tables, Figures, Formulas, Captions, software mentions
Software: Dependency Data and Risk, Repo activity history, analysis of domain software, research workflow objects
Context eg. "What role is a chemical used for?"
More complex concepts: assays, endpoints, exposures
{{embed: 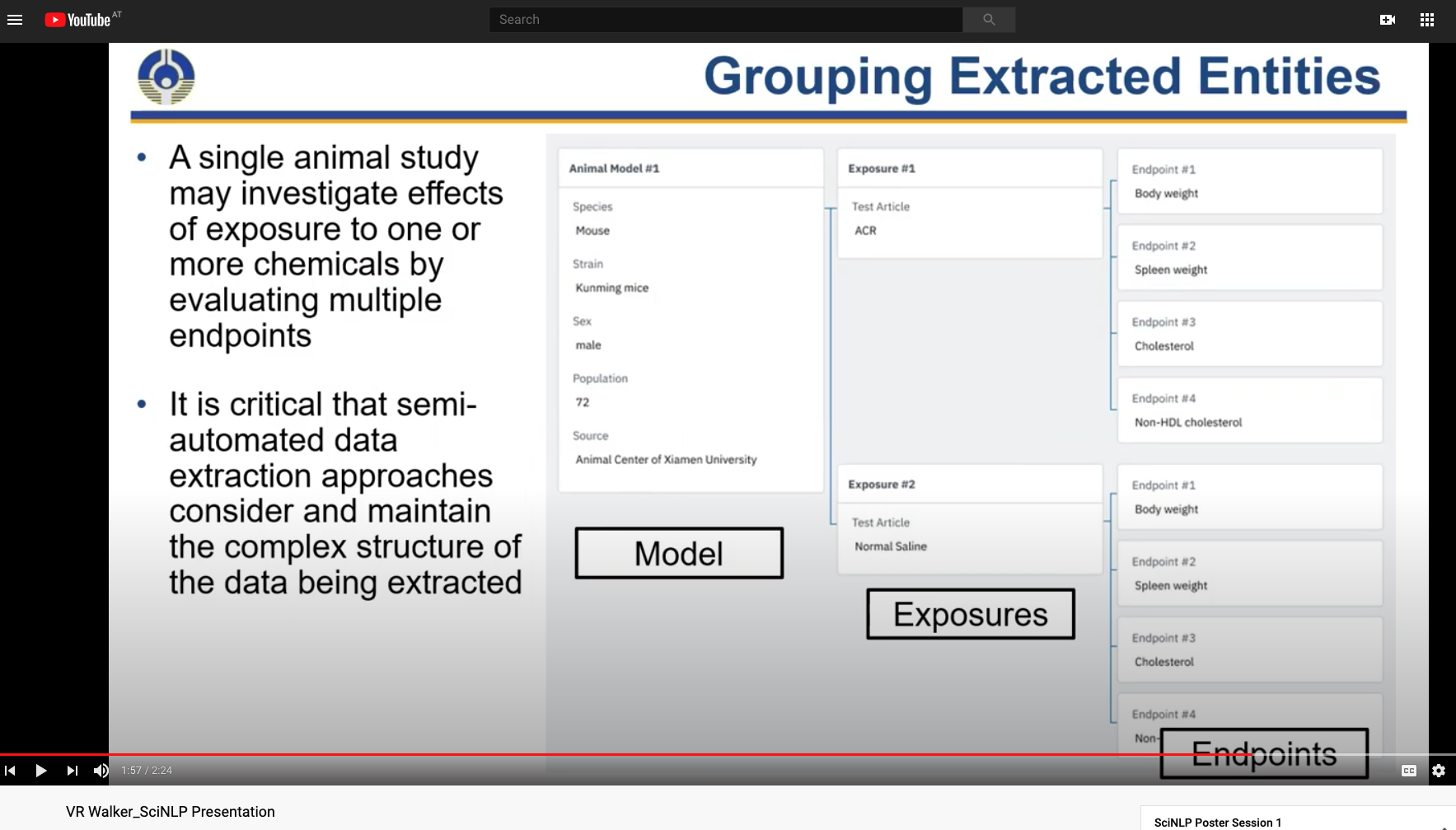
Exciting Developments
SPECTER: scientific document level embeddings based on citations and transformers
SNORKEL and FLYINGSQUID -- HazyLab
https://www.algolia.com/ Better Search
open, big questions
What effects will GPT-4 have on the tasks? Can the generator be flipped as a discriminator? Can parsing rules be generated?
As most of the pipeline becomes neural and through Dependency Tree-> dependency forest (tree -into-> 3D tensor), how will the workflow change?
INDRA Havard Team. Looks insanely advanced
INDRA (the Integrated Network and Dynamical Reasoning Assembler) assembles information about biochemical mechanisms into a common format that can be used to build several different kinds of explanatory models. Sources of mechanistic information include pathway databases, natural language descriptions of mechanisms by human curators, and findings extracted from the literature by text mining. Mechanistic information from multiple sources is de-duplicated, standardized and assembled into sets of mechanistic Statements with associated evidence. Sets of Statements can then be used to assemble both executable rule-based models (using PySB) and a variety of different types of network models.
INDRA-IPM also recognizes protein families and complexes and grounds them in the FamPlex ontology. In some cases, there is ambiguity in the name of a specific gene and a family it is part of. An example of this is the grounding of “JUN” from text to the JUN family, which also includes the JUN gene. In this case the user can use a synonym such as “c-JUN” that refers to the singular entity in order to reference only the gene and not the family.
We have exposed two reading systems to users. The REACH reader developed by the CLU Lab at the University of Arizona is an information extraction system for the biomedical domain, which aims to read scientific literature and extract cancer signaling pathways. We recommend users try REACH first due to its speed. The TRIPS/DRUM system developed by IHMC may offer greater mechanistic detail in some use cases (for instance, it supports recognizing complex molecular conditions such as “BRAF-V600E not bound to Vemurafenib”), but it requires significantly longer to run.

literature based discovery is integrated with system biology path analysis. Click an edge and you see where the relation is from
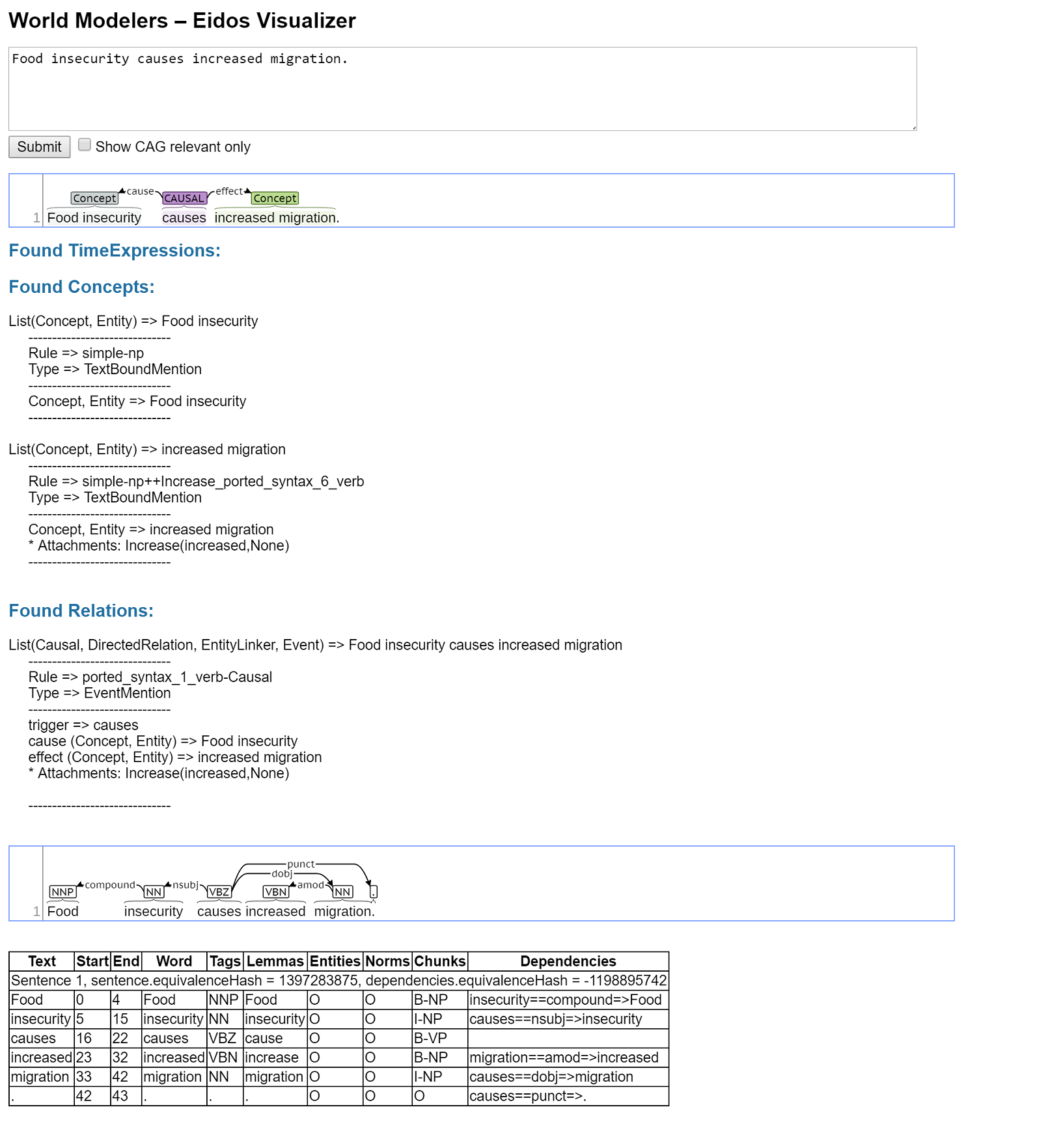
Eidos is an open-domain machine reading system designed by the Computational Language Understanding (CLU) Lab at University of Arizona for the World Modelers DARPA program. It uses a cascade of Odin grammars to extract events from free text.
[Odin][rule based system | RBS]] information extraction
This is huge, because you can interactively develop Dependency Tree Search Patterns in Research and debug in realtime
RuleBender. RuleBased (~agent-based) modeling and visualization. system biology bioinformatics
Task analysis
There are two broad motivations for comparing the similarities and differences within a family of models. In the first case, a research team is building a family of models up from a base model over time. As members leave the project, new members join to replace them. The continuity of the project is thus greatly facilitated by the ability of the new members to browse the history of the model and identify when and where modifications were made. Identifying the common core among the family of models is essential, since the elements that are not present in the core represent modifications to the model.
In the second case, a researcher intends to model a particular signaling pathway or set of pathways. As part of this process, they would want to see what elements of that pathway have been previously modeled, and explore the relationships among existing models in the literature. The researcher downloads several models from one of the several existing online databases [14–17] in a commonly-used model exchange format such as the Systems Biology Markup Language (SBML) [18]. The researcher would like to see at a glance which model components are shared and which are unique.
Starting from these two motivating cases, and through close interaction with domain experts, we identified the following major tasks where visualizations can benefit model comparison in the area of cell signaling. Because of the similarities between model usage in this domain and in other domains, we assert that many of these tasks have global applications to model comparison beyond the cell signaling domain.
Identify similar structures within models. Identifying similar structures is beneficial because if two different models share a common core, it is likely that those models can be combined to form a single, more-complete model. Additionally, searching for a single structure common to a significant subset of a family of models can help to identify models missing this structure. This can help researchers make observations about the functionality of that subset of models.
Identify structures that differ between pairs of models. Performing a pairwise comparison similar to task 1 with the goal of identifying structures that differ between the models helps researchers identify model components present in one model that do not appear in the other. Researchers can use this information to explore the functional effects of the structural differences between models. When identifying both the similarities and differences between graphs, minimizing layout differences is essential to enable the user to see changes [19, 20].
Sort/cluster models by similarity. Sorting models by degree of similarity helps to minimize visual differences between graphs in proximity to each other, facilitating comparison [19]. As such, a method for computing the similarity of a pair of models should be developed or found from literature. Following this, the models should be laid out based on these scores in a clear and visually pleasing way.
Support pairwise detailed comparison. Building upon the similarity and difference comparison of a pair of models, a researcher should also be able to examine the similar or differing structures of the models in more detail. In particular, the researcher may wish to examine the individual rules within the model to determine the level of similarity.
Explore the functional effects of differences between model structures. The researcher may also wish to explore the functional effects of model changes. In particular, the researcher should be able to perform a pairwise comparison of the simulation results or other species and reactions in the generated network of a model, in order to identify how the changes within a model affect the generated outputs.
Organize and browse model repositories. A researcher should be able to use this system to organize and browse a set of possibly unrelated models from a database or online repository. The researcher should still be able to look at the similar and different structures across the collection of models under examination.
Enable the ability to share model layouts with other researchers. Finally, if a researcher wishes to highlight important structural features that were custom-encoded into a model, that researcher must be able to also convey the structure of the model along with the model itself. To keep the model interactive and to share all of the properties of the model, simply sharing a screenshot of a model is not sufficient. Therefore, although the model language may not specify any kind of set structural information, that structural information needs to be maintained.
This task analysis breakdown shows that a number of problems related to the comparison of models can be solved or aided with visualization. Specifically, Tasks 1–6 can be performed with a clear visual representation of the model(s), and are specifically addressed in this work. Task 7, on the other hand, is not specifically a visualization challenge, but can be facilitated by specific aspects of our visualization system.
Cytoscape enables network visualization and provides access to a large ecosystem of analysis plugins; more information is available at: https://cytoscape.org/cy3.html
dataset/bio/description::
dataset/bio/comment::
Kappa Language Harvard
The challenge of building models of complex biomolecular systems necessitates an intermediate stage to bridge between the biophysically and biochemically grounded descriptions in papers and databases on the one hand and the ungrounded abstract language of Kappa on the other. This intermediate staging area is a knowledge representation that enables the user-driven aggregation of nuggets encapsulating mechanistic information pertaining to protein-protein interactions, the visualization of their interrelations, identification of conflicts, etc. In essence, it aims at being the biologically grounded model the user reasons about. This model is then compiled into Kappa for execution. From this perspective, Kappa is seen as an "assembler" code rather than the primary language for building a model in the first place.
KAMI stands for Knowledge Aggregator and Model Instantiator. It is an ongoing development led by Russ Harmer at ENS Lyon. KAMI poses interesting challenges in knowledge representation and multi-level graph rewriting.
New ACL Papers 2020
BioPortal Search for BioOntologies
BioDati Kinda unusable but integrates nanopubs and Biological Expression Language (BEL). You can lasso clusters in the biological network etc.
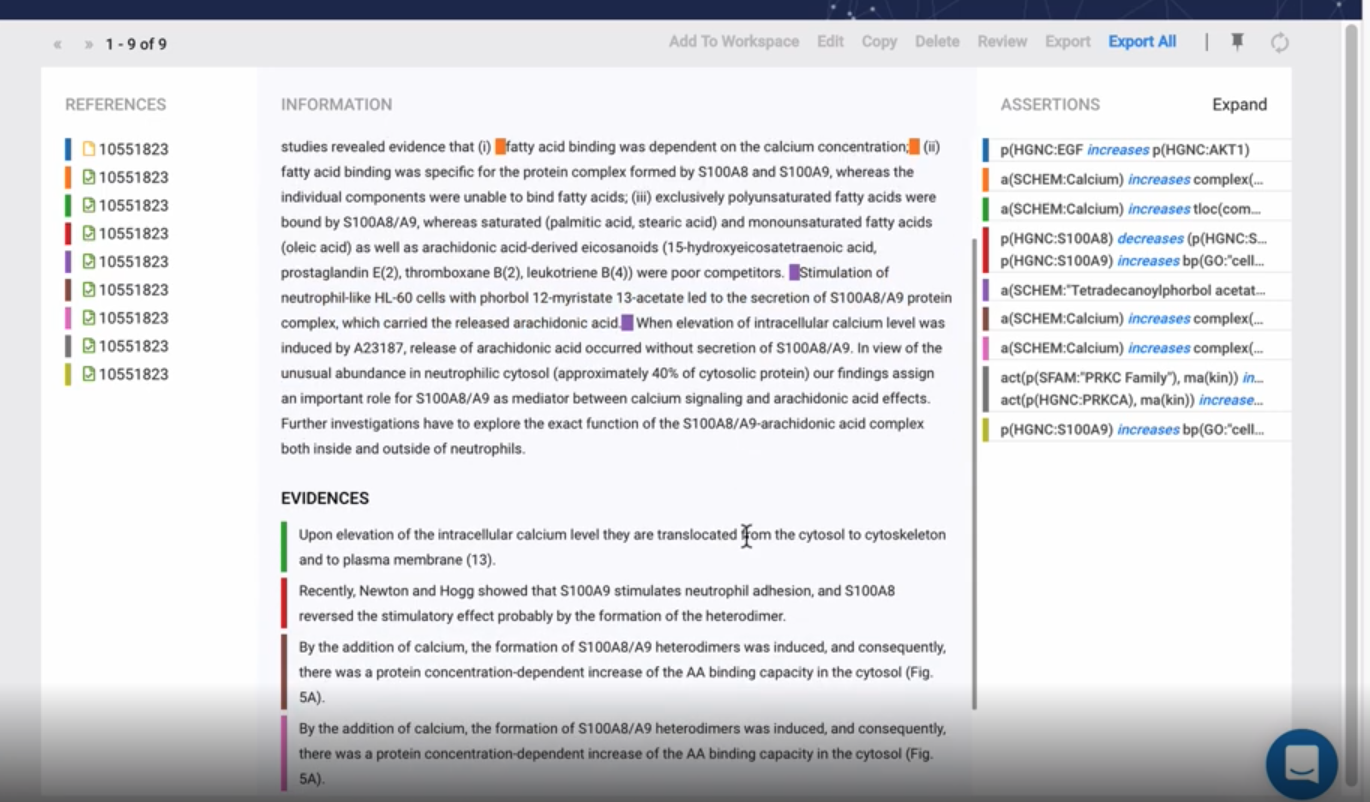
Nanopubs
[[interval:100.0]] [[factor:2.20]] April 11th, 2021
Nanopublications are implemented in the language RDF and come with an evolving ecosystem of tools and systems. They can be published to a decentralized server network, for example, and then queried, accessed, reused, and linked.
Furthermore, because nanopublications can be attributed and cited, they provide incentives for researchers to make their data available in standard formats that drive data accessibility and interoperability. Nanopublications have the following general structure:

As can be seen in this image, a nanopublication has three basic elements:
Learn from these Guys PhysioNet
DSL for expressing biological findings Extracting Meaning from Scientific Text
spaCy’s parser component can be used to be trained to predict any type of tree structure over your input text – including semantic relations that are not syntactic dependencies. This can be useful to for conversational applications, which need to predict trees over whole documents or chat logs, with connections between the sentence roots used to annotate discourse structure.
For example, you can train spaCy’s parser to label intents and their targets, like attributes, quality, time and locations. The result could look like this:

UNCURL-App is a unified framework for interactively analyzing single-cell RNA-seq data. It is based on UNCURL for data preprocessing and clustering. It can be used to perform a variety of tasks such as:
Unsupervised or semi-supervised preprocessing
Clustering
Dimensionality reduction
Differential expression
Interactive data analysis and visualization
[[interval:40.4]] [[factor:2.95]] September 25th, 2020
dataset/bio/description::
Reactome is a free, open-source, curated and peer-reviewed pathway database.
dataset/bio/comment::
https://biothings.io/ Annotation as a Service (variants, genes)

QTL Table Miner. Mining relations from structured plots
We present QTLTableMiner++ (QTM), a table mining tool that extracts and semantically annotates QTL information buried in (heterogeneous) tables of plant science literature
A significant amount of experimental information about Quantitative Trait Locus (QTL) studies are described in (heterogenous) tables of scientific articles. Briefly, a QTL is a genomic region that correlates with a trait of interest (phenotype). QTM is a command-line tool to retrieve and semantically annotate results obtained from QTL mapping experiments.
Mining gene pathway figures :app with OCR for figures
In this study, we aimed to identify pathway figures published in the past 25 years, to characterize the human gene content in figures by optical character recognition, and to describe their utility as a resource for pathway knowledge.
[...] challenge would be to link data across multiple biological entities. For example, fields like systems chemical biology, which studies the effect of drugs on the whole biological system, requires the integration and cross-linking of data across from multiple domains, including genes, pathways, drugs as well as diseases
meta.org Research feeds with context (Chan Zuckerberg initiative)
embedding structural dependency?
Visualizations and Representations
Bivariate causal discovery: Bablyon Health ... not sure how legit they are
Snorkel and data programming as a way to generate more training data from heuristic functions
Distant Supervision, but with less noise
Polyglot Programming Environments: Imagine Clojure powers with python machine learning ecosystem
SemRegex or semgrex: Regex but semantically applied over a syntax Dependency Tree,
SemRegex provides solutions for a subtask of the program synthesis problem: generating regular expressions from natural language. Different from the existing syntax-based approaches, SemRegex trains the model by maximizing the expected semantic correctness of the generated regular expressions.
The semantic correctness is measured using the DFA-equivalence oracle, random test cases, and distinguishing test cases. The experiments on three public datasets demonstrate the superiority of SemRegex over the existing state-of-the-art approaches.
measurement benchmark programming language

{kind=link}
Stanford Core NLP in idiomatic Clojure! in Clojure - probably possible through Java interoperability
New approaches to approximate causality from correlation
"Correlation does not equal Causation" might be an unproductive trope similar to "If you think you understand quantum physics, you don't understand quantum physics"
Kite Language Support for Python in JupyterLab
The goal of probabilistic programming is to enable probabilistic modeling and machine learning to be accessible to the working programmer, who has sufficient domain expertise, but perhaps not enough expertise in probability theory or machine learning. We wish to hide the details of inference inside the compiler and run-time
BioGrakn ¨b****is a collection of knowledge graphs of biomedical data demonstrating the following use-cases:
- Text Miningtext_miningPubMed
- BLASTblastN/A
- Disease Networkdisease_networkUniprot, Reactome, DGIdb, DisGeNET, HPA-Tissue, EBI IntAct, Kaneko, Gene Expression Omnibus and TissueNet
Entity Types
diseases, drugs, genes, variants, proteins, pathways